项目名称:(RNA-seq)转录组测序及分析报告
所属分类:生物信息学分析-报告解读
联系电话:020-85625352
QQ:386244141
Email:servers@gzscbio.com
转录组测序及分析报告
生信部
2021年03月19日
项目信息
合同编号:xx-xx-202x-xx-xx
客户姓名:xxx
客户单位:xxxxxx
1. 分析流程
1.1. 建库测序流程
从RNA样品提取到最终数据获得,样品检测、建库、测序等每一环节都会直接影响数据的数量和质量,从而影响后续数据分析的结果。为从源头保证测序数据准确可靠,在数据的所有生产环节都严格把关,从根源上确保高质量数据的产出。建库测序的流程:
Total RNA 样本检测
RNA 富集
双链cDNA合成
末端修复、加A和接头
片段选择和 PCR 扩增
文库质检
Illumina测序
1.2. 信息分析流程
RNA-seq的核心是基因表达差异的显著性分析,使用统计学方法,比较两个条件或多个条件下的基因表达差异,从中找出与条件相关的特异性基因,然后进一步分析这些特异性基因的生物学意义,分析过程包括质控、比对、定量、差异显著性分析、功能富集等环节。信息分析流程如下图所示:
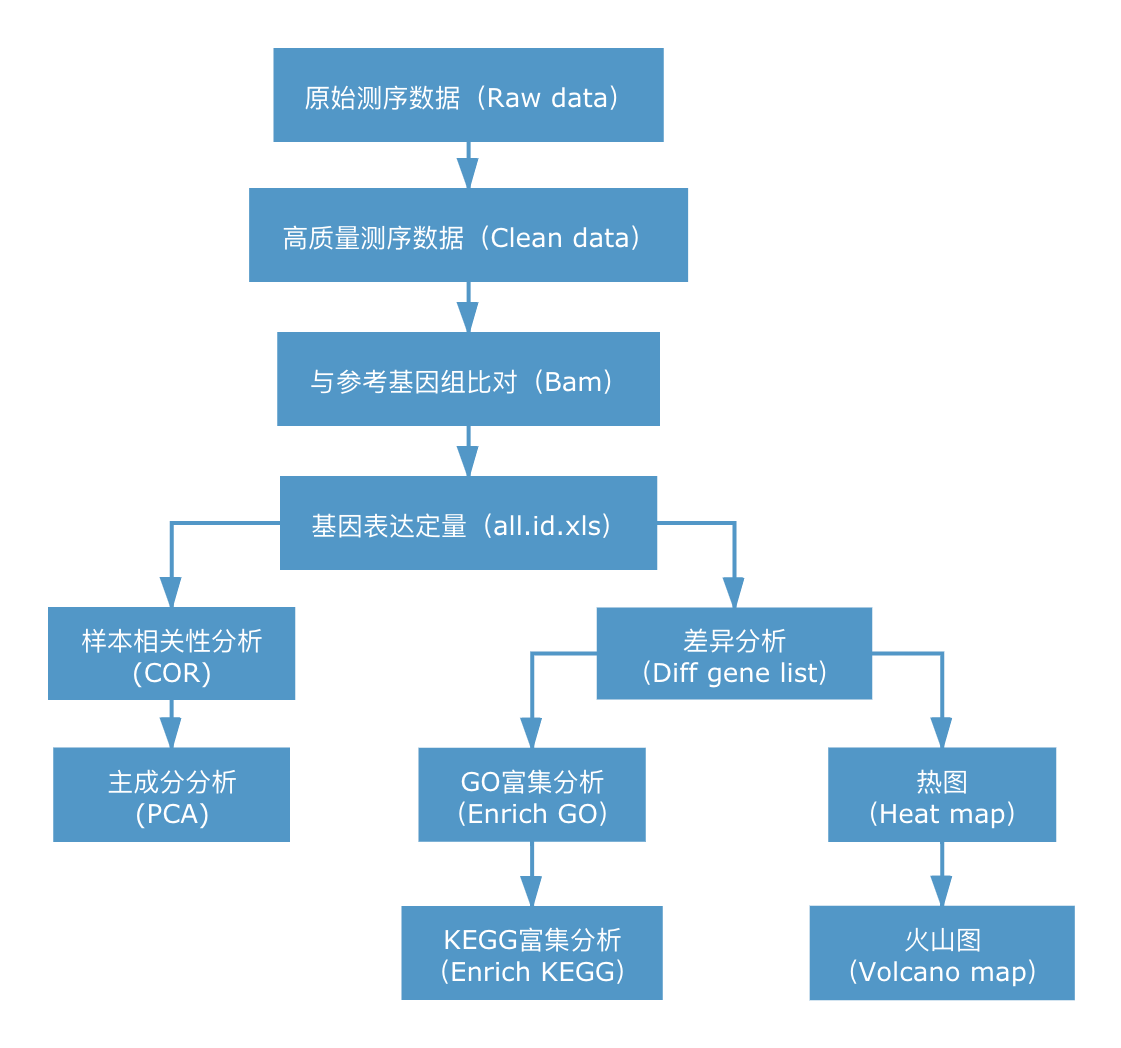
2. 信息分析
2.1. 测序数据质量控制
对原始测序数据及去除接头后的可用数据进行质量评估。测序数据一般为双端测序,因此,每个测序样本会有两个测序结果。
评估的具体内容见:
RawData-fastqc 文件链接: /result/qc/qc_rawdata/*.html
CleanData-fastqc 文件链接: /result/qc/qc_cleandata/*.html
Fastqc 格式补充说明: /result/qc/qc_Supplement.html
2.2. 参考基因组比对
测序片段(fragments)是mRNA随机打断的,为了确定这些一段由哪些基因转录来,需要将质控后的clean reads比对到参考基因组上。使用HISAT2软件将Clean Reads与参考基因组进行快速精确的比对,获取Reads在参考基因组上的定位信息[4]。HISAT2软件官方手册。
如果参考基因组组装的较为完善,而且所测物种与参考基因组一致,且相关实验不存在污染,那么实验所产生的测序reads成功比对到基因组的比例会高于70% (Total Mapped Reads or Fragments)。本项目所用参考基因组为 hg38 ,下载链接:Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz,基因组结构注释文件:Homo_sapiens.GRCh38.90.gtf.gz。
结果文件:
各个样本的比对情况统计文件:
/result/map_stat/*.flagstat.txt
2.3. 定量分析
2.3.1. 基因表达定量
我们对每个样本分别进行基因表达水平的定量分析,再合并得到所有样本的表达矩阵,第一列为基因的ID,其余列为各样本的原始read count值,seqname列之后为该基因注释信息。
表格说明:
| 表头 | 说明 |
|---|---|
Geneid | 基因名 |
Samples_name* | 样本的表达矩阵原始read count值 |
... | 同上 |
seqname | 基因所在的染色体名称 |
start | 基因所在染色体的起始位置 |
end | 基因所在染色体的终止位置 |
strand | 基因所在染色体的正负链信息 |
ENSEMBL | 基因名称ENSEMBL |
SYMBOL | 基因名称SYMBOL |
biotype | 基因注释中对应的biotype |
description | 基因功能描述 |
结果文件:
原始表达矩阵及注释结果:
result/Quant/gene_counts.xls
2.3.2. 样本间相关性
生物学重复通常是任何生物学实验所必须的,目前主流期刊也基本要求生物学重复。生物学重复主要有两个用途:一个是证明所涉及的生物学实验操作不是偶然,而是可重复的。另一个是为了确保后续的差异基因分析得到更可靠的结果。样品间基因表达水平相关性是检验实验可靠性和样本选择是否合理的重要指标。相关系数越接近1,表明样品之间表达模式的相似度越高。Encode计划建议皮尔逊相关系数的平方(R2)大于0.92(理想的取样和实验条件下)。具体的项目操作中,我们要求生物学重复样品间R2至少要大于0.8,否则需要对样品做出合适的解释,或者重新进行实验。根据各样本所有基因的表达值计算组内及组间样本的相关性系数,绘制成热图,可直观显示组间样本差异及组内样本重复情况。样本间相关性系数越高,其表达模式越为接近,样本相关性热图如下图所示。
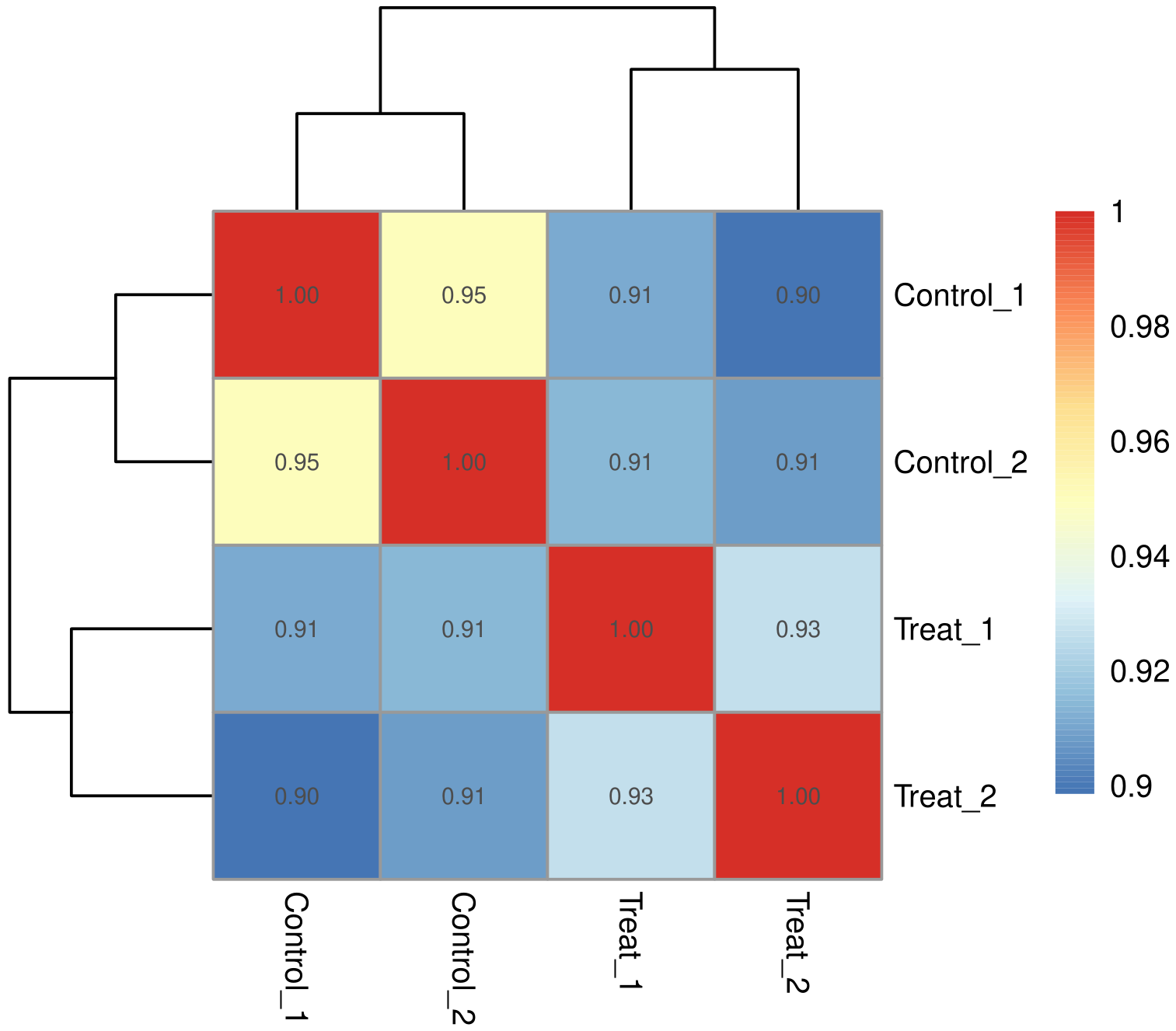
图 1 样本间相关性热图
图中横纵坐标为各样本相关系数的平方
结果文件:
样本间相关性热图结果:Quant/cor_pheatmap*
2.3.3. 主成分分析
主成分分析(PCA)也常用来评估组间差异及组内样本重复情况,PCA采用线性代数的计算方法,对数以万计的基因变量进行降维及主成分提取。我们对所有样本的基因表达值进行PCA分析,如下图所示。理想条件下,PCA图中,组间样本应该分散,组内样本应该聚在一起。
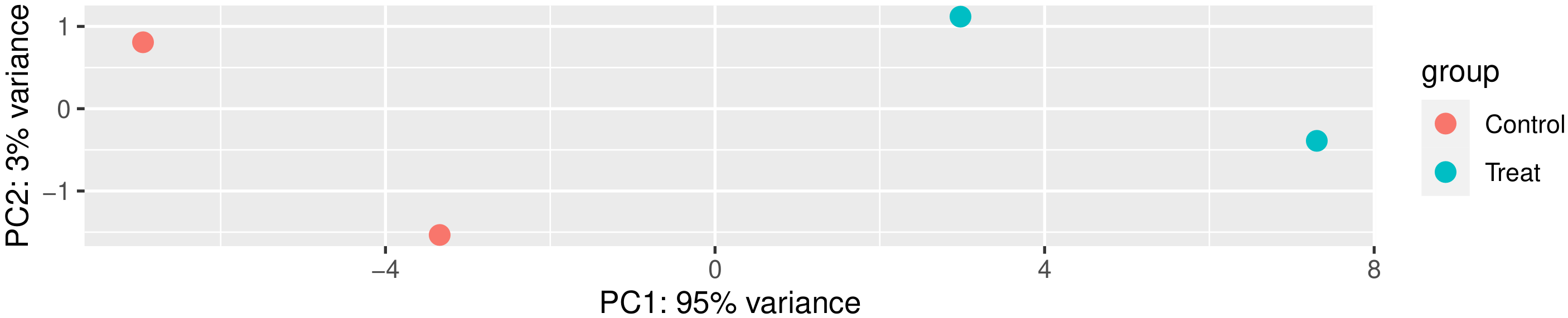
图 2 主成分分析结果图
图中横坐标为第一主成分,纵坐标为第二主成分
结果文件:
主成分分析结果:Quant/pca*
2.4. 差异分析
基因表达定量完成后,需要对其表达数据进行统计学分析,筛选样本在不同状态下表达水平显著差异的基因。差异分析主要分为三个步骤。
首先对原始的readcount进行标准化(normalization),主要是对测序深度的校正。
然后统计学模型进行假设检验概率(pvalue)的计算
最后进行多重假设检验校正,得到FDR值(错误发现率,padj是其常见形式)[1-2]。
针对不同的实验情况,我们选用合适的软件进行基因表达差异显著性分析,具体如下表所示。
表1 表达差异分析所用软件及差异基因筛选标准
| 类型 | 软件 | 标准化方法 | pvalue计算模型 | FDR计算方法 | 差异基因筛选标准 |
|---|---|---|---|---|---|
| 有生物学重复 | DESeq2(Anders et al, 2014) | DESeq | 负二项分布 | BH | |log2(FoldChange)| > 0 & padj < 0.05 |
| 无生物学重复 | edgeR(Robinson et al, 2010) | TMM | 负二项分布 | BH | |log2(FoldChange)| > 1 & padj < 0.05 |
若按照以上标准筛选得到的差异基因过少(低于100),很有可能导致后面的功能富集分析没有显著性结果,所以,我们会根据项目的具体情况,适当地降低筛选差异基因的阈值标准。若项目实验只关注某几个基因的表达情况(如基因敲除),不在意富集结果,从下面的差异分析表格中筛选关注的那几个基因即可。
一般来说,如果一个基因在两组样品中的表达量差异达到两倍以上,我们认为这样的基因是具有表达差异的。为了判断两个样品之间的表达量差异究竟是由于各种误差导致的还是本质差异,我们需要对所有基因在这两个样本中的表达量数据进行假设检验。而转录组分析是针对成千上万个基因进行的,这样会导致假阳性的累积,基因数目越多,假设检验的假阳性累积程度会越高,所以引入padj对假设检验的P-value进行校正,从而控制假阳性的比例[3]。
差异基因的筛选标准是非常重要的,我们给出的标准|log2(FoldChange)| > 1 & padj< 0.05是常用的经验值,在实际项目中可以根据情况灵活选择。例如,差异倍数可以选择1.5倍,也可以选择3倍,padj常用的阈值包括0.01、0.05、0.1等。若按照以上标准筛选得到的差异基因过少,很有可能导致后⾯的功能富集分析没有显著性结果。若项目实验只关注某几个基因的表达情况(如基因敲除),不在意富集结果,从下面的差异分析表格中筛选关注的那几个基因即可。反之,如果得到的差异基因数目过多,不利于后续目标基因的筛选,这个时候可使用更严格的阈值标准进行筛选,则可以使用更严格的阈值标准进行筛选。
2.4.1. 差异基因的筛选
通过Deseq2进行差异分析,我们通常采用 |log2FC|>1 & padj < 0.05 进行差异基因的筛选,随后对差异基因进行注释,得到包含注释信息的差异基因列表。
结果文件:
差异基因列表及相关注释信息(筛选结果):result/Enrichment/Allgene_anno.xls
差异基因列表及相关注释信息(总的结果):result/Enrichment/Allgene_anno_ALL.xls
Differential/Allgene_anno*.xls表头
Differential/Allgene_anno*.xls表头
Differential/Allgene_anno*.xls表头
Differential/Allgene_anno*.xls表头
Differential/Allgene_anno*.xls表头
Differential/Allgene_anno*.xls表头
Differential/Allgene_anno*.xls表头