项目名称:micRNA测序与分析报告
所属分类:生物信息学分析-报告解读
联系电话:020-85625352
QQ:386244141
Email:servers@gzscbio.com
micRNA测序与分析报告
1. 建库测序工作流程
1.1. 建库测序流程
MicroRNA (miRNA) 是一类长度约为20-24个核苷酸长度的具有调控功能的非编码RNA,miRNA 是生物体内一类重要的特殊分子,诱导基因沉默,参与细胞生长、发育、基因转录和翻译等诸多生命活动的调控过程。
从RNA样品到最终分析结果数据,需要经过样品检测、建库、测序和信息分析等过程,其中测序过程我们采用高通量测序illumina HiSeq/MiSeq测序平台;illumina测序得到的原始图像数据文件经碱基识别(Base Calling)分析转化为原始测序序列(Sequenced Reads),我们称之为Raw Data或Raw Reads,结果以FASTQ(简称为fq)文件格式存储,其中包含测序序列(reads)的序列信息以及其对应的测序质量信息。
样品质量检测:利用NanoDrop 2000分光光度计对RNA样品进行初步定量,Aglient 2100对样品浓度精确定量。RNA样品总量、浓度、完整性(RIN值,RNA Integrity Number)、纯度(OD260/OD280比值)符合收样立项标准后,进入下一步的文库构建。
文库构建:Total RNA PEG (polyethylene glycol)沉淀,连接3’接头,PAGE (polyacrylamide gel electrophoresis) 胶分离回收,连接5’接头,反转录,PCR扩增,文库切胶回收。
文库质检:Q-PCR方法对文库进行精确定量,文库有效浓度>2nM即可上机测序。
上机测序:将各文库按照有效浓度及目标下机数据量的需求pooling后,进行SE75测序。

图1: Small RNA 文库构建 Workflow
Small RNA测序的接头信息:
RNA 5‘Adapter (RA5), part:
5’-GTTCAGAGTTCTACAGTCCGACGATC-3’
RNA 3’Adapter (RA3), part:
5’-AGATCGGAAGAGCACACGTCT-3’ (1ug流程)
5’-CTGTAGGCACCATCAATCAGATCGGAAGAGCACACGTCTG-3’ (10ug流程)
2. 信息分析流程
Raw Data通过去接头、去低质量、去污染、统计序列长度分布等过程完成初级分析;将初级分析得到的clean reads与ncRNA库比对,分类注释ncRNA,并去除reads中的rNRA、tRNA、snRNA、snoRNA等;然后再将reads比对到参考基因组、miRBase上,计算miRNA表达量,注释已知miRNA、预测新的miRNA;然后进行差异miRNA、靶基因预测、功能注释等后续分析。
miRNA生物信息分析流程图

2.1. miRNA-seq 测序数据质量评估
2.1.1. 数据质量评估 及 数据过滤
对于测序得到的rawdata,我们首先使用fastqc进行数据质量评估,通过结果,我们可以了解到测序数据reads质量、长度、碱基分布,序列重复频次等基本信息。
测序得到的raw data,里面含有带接头的、低质量的reads,为了保证后续信息分析的质量,必须对raw reads进行处理,得到clean reads。
raw data的过滤步骤如下: 去除低质量 reads; 去除有5’接头污染的reads; 去除没有3’接头序列; 去除没有插入片段的reads; 去除长度小于16nt的reads;
数据经过以上过滤后,我们对数据做一些基本统计。
结果:
| 结果说明 | 结果文件 |
|---|---|
| 原始数据 RawData QC 结果 | 00.QC/qc_rawdata/ |
| 过滤数据 CleanData QC 结果 | 00.QC/qc_cleandata |
| Trimmed Reads (unique) 结果文件 | 00.QC/*.trim.collapse.fa |
2.2. 数据长度分布
一般来说,小RNA的长度区间为18~30nt,数据过滤完成后,我们统计了clean reads数目及各自占总reads的百分比,并绘制reads长度分布图。reads长度分布图能帮助我们判断小RNA的种类,如miRNA集中在21或22nt,siRNA集中在24nt,piRNA集中在30nt。
Clean Data Reads 长度分布统计直方图:




Figure 2.2: Small RNA Clean Reads长度分布图
Clean Reads长度分布直方图,横坐标为Reads的长度,纵坐标为样品中对应该长度的total Reads数量。
2.3. miRNA基因组比对分析
我们将Clean Reads 与 mature miRNA, miRNA hairpin, mRNA, mature & primary tRNA, snoRNA, rRNA, other non-coding RNA, and (optional) known RNA 使用bowtie进行比对搜索,然后统计reads的比对情况,分析样本中各类ncRNA的数目及比例情况,统计结果如下。过滤出待分析的miRNA以便后续分析,将clean reads中ncRNA尽可能地去除,便于后续 miRNA 检测及预测。
Reads类型分布图

图显示的是各类型的RNA reads和miRNA reads占总distinct reads的比例。
与参考基因组比对质控结果:
| 结果说明 | 结果文件 |
|---|---|
| Trimmed Reads (unique) 与参考基因组的比对结果(bam) | 01.Mapping/*.bam |
| Trimmed Reads (unique) 与参考基因组的比对结果(bw) | 01.Mapping/*.bw |
| 各类型RNA统计情况 | 00.QC/annotation.report.csv |
| 与各个smallRNA比对的详细结果 | 01.Mapping/mapped.csv |
2.4. 已知 miRNA 的检测以及 novel miRNA 预测分析
首先用去除了ncRNA后的Reads比对miRbase数据库,比对上miRbase中 reads 用于检测已知成熟的miRNA,并统计其表达量和RPM值。我们采用miRge软件来进行 miRNA reads 检测注释、novel miRNA的预测、miRNA表达量的统计。
miRNA分析结果汇总:
| 结果说明 | 结果文件 |
|---|---|
| 基本注释结果: | |
| miRNA reads 变体类型注释结果 | 02.miRNA/1.isomiR/sample_miRge3.gff |
| miRNA reads 变体类型可视化统计 | 02.miRNA/1.isomiR/*svg |
| miRNA碱基编辑分析结果: | |
| A2I在各个样本中统计汇总表 | 02.miRNA/2.a2IEditing/a2IEditing.report.csv |
| A2I在各个样本中统计汇总表的筛选及简化信息 ( 即汇总表的 *.RPM.mismatch、*.AtoI.percentage列 ) | 02.miRNA/2.a2IEditing/a2IEditing.report.newform.csv |
| A2I分析的序列及统计详情信息 | 02.miRNA/2.a2IEditing/a2IEditing.detail.txt |
| 二级结构预测结果: | |
| novel miRNA 二级结构预测结果 | 02.miRNA/3.novel_predict/*.R1_novel_miRNAs/ |
| novel miRNA 二级结构预测详细信息表格汇总 | 02.miRNA/3.novel_predict/*.R1_novel_miRNAs/*_novel_miRNAs_report.csv |
以上miRNA各图的可视化网页:02.miRNA/miR_visualization.html
2.4.1. miRNA异构体分析
随着高通量测序技术的发展,早期认为的miRNA loci仅产生1条miRNA成熟体序列这一观点被颠覆。对于某一miRNA来说,它并不是单一的序列,而是由一系列长度/序列及表达不同的异构体 (isomiRs) 组成。这些isomiRs表达多样且序列多样,甚至引入多样的5'端及种子区域。特定miRNA位点在疾病组织中可具有异常的表达模式,现已证实,部分isomiRs具有重要的生物学功能。
IsomiRs目前主要分为三类:5′isomiRs, 3′isomiRs, polymorphic isomiRs。IsomiRs的产生机制主要有:miRNA加工和成熟过程中Darsha和Dicer酶的不精确或选择性剪切;3'端核苷酸添加;RNA编辑和单核苷酸多态性( single nucleotide polymorphism,SNP)。主要表现为:5'端修剪;3' 端修剪;3'端核苷酸附加和碱基替换。其中,5'端修剪和碱基替换可发生在种子区域内部,产生“种子转移”(seed shifting)。
我们通过序列与miRbase比对的情况,将各个miRNA Reads进行isomiR识别,并同时汇总至miRNA reads 注释结果中,分类统计,并选取表达量较高的前20个mirna进行各类IsomiRs表达丰度统计。

图2.4.1 所有样本的变体类型分布(isomiRs)




图2.4.2 各个样本的变体分布TOP20的丰度统计
2.4.2. miRNA 碱基编辑
成熟miRNA序列的第2-8个碱基被称作“种子”序列,保守性很高。若在这一区域发生碱基突变,则可能改变miRNA的靶基因作用位点。我们通过将miRNA序列与miRBase中已知miRNA前体以及成熟miRNA序列进行比对(只允许一个位点的错配)找出发生了碱基突变的miRNA,统计汇总在该miRNA的碱基突变位点,以及发生碱基编辑的Reads数量及百分比,并对统计概况结果进行可视化。

图2.4.3 鉴定为已存在mirna碱基编辑的在各个样本中的相应readCount占比统计图
miRNA碱基编辑分析结果详细说明:
A2I在各个样本中统计汇总表
表头 说明 miRNAmiRNA的名称 A-to-I position in the miRNAA2I检测位置,即序列的第几个碱基 miRNA sequencemiRNA的序列 Sample.readCount该样本miRNA的所有序列的readCount总和 Sample.readCount.canonical该样本miRNA的所有序列的readCount总和(筛选低质量后) Sample.RPM.canonical该样本miRNA的所有序列的readCount总和(筛选低质量后)的RPM值 Sample.readCount.mismatchA2I检测的序列readCount总和 Sample.RPM.mismatchA2I检测的序列readCount总和的RPM值 Sample.AtoI.percentageA2I检测的序列readCount占比,计算方法为: Sample.readCount.mismatch/Sample.readCount.canonical*100%Sample.AtoI.adjusted.pValueA2I检测的置信度计算 A2I在各个样本中统计汇总表的筛选及简化信息
( 即汇总表的*.RPM.mismatch、*.AtoI.percentage列,该表对应上面的可视化结果 )表头 说明 miRNA:positionmiRNA和position位置信息 sample样本名 A-to-I percentageA2I检测的序列readCount占比,对应第一个表中的 Sample.AtoI.percentagelog2RPMA2I检测的序列readCount总和的log2RPM值,对应第一个表中的A2I检测的序列
readCount总和的RPM值Sample.RPM.mismatch,进行log2计算A2I分析的序列及统计详情信息
每一个miRNA块,包含3小块内容:原始序列、筛选后的序列统计、筛选后的序列。重复的miRNA块代表不同样本,顺序为 A2I在各个样本中统计汇总表 的样本信息。
每一行序列包含3个信息,
miRNA sequence(reads序列),*.readCount(Count计数),Filter(是否筛选到后续进行碱基编辑分析)
2.4.3. novel miRNA 预测及二级结构分析
miRge根据序列特征,碱基配对原则,最小自由能等特征模型,进行新颖的miRNA及二级结构预测。预测示意图如下。结果见:02.miRNA/3.novel_predict/

图2.4.4 新miRNA的二级结构预测示意图
2.5. miRNA 定量分析
2.5.1. 样本间相关性分析
生物学重复通常是任何生物学实验所必须的,目前主流期刊也基本要求生物学重复。生物学重复主要有两个用途:一个是证明所涉及的生物学实验操作不是偶然,而是可重复的。另一个是为了确保后续的差异分析得到更可靠的结果。样品间基因表达水平相关性是检验实验可靠性和样本选择是否合理的重要指标。相关系数越接近1,表明样品之间表达模式的相似度越高。Encode计划建议皮尔逊相关系数的平方(R2)大于0.92(理想的取样和实验条件下)。具体的项目操作中,我们要求生物学重复样品间R2至少要大于0.8,否则需要对样品做出合适的解释,或者重新进行实验。根据各样本所有基因的表达值计算组内及组间样本的相关性系数,绘制成热图,可直观显示组间样本差异及组内样本重复情况。样本间相关性系数越高,其表达模式越为接近,样本相关性热图如下图所示。

图 2.5.1. 样本间相关性热图
图中横纵坐标为各样本相关系数的平方
结果文件:
样本间相关性热图结果:
04.DE/Quant/cor_pheatmap*
2.5.2. 主成分分析
主成分分析(PCA)也常用来评估组间差异及组内样本重复情况,PCA采用线性代数的计算方法,对数以万计的基因变量进行降维及主成分提取。我们对所有样本的基因表达值进行PCA分析,如下图所示。理想条件下,PCA图中,组间样本应该分散,组内样本应该聚在一起。

图 2.5.2. 主成分分析结果图
图中横坐标为第一主成分,纵坐标为第二主成分
结果文件:
主成分分析结果:
04.DE/Quant/pca*
2.6. miRNA 差异分析
基因表达定量完成后,需要对其表达数据进行统计学分析,筛选样本在不同状态下表达水平显著差异的基因。差异分析主要分为三个步骤。
首先对原始的readcount进行标准化(normalization),主要是对测序深度的校正。
然后统计学模型进行假设检验概率(pvalue)的计算
最后进行多重假设检验校正,得到FDR值(错误发现率,padj是其常见形式)[1-2]。
针对不同的实验情况,我们选用合适的软件进行基因表达差异显著性分析,具体如下表所示。
表1 表达差异分析所用软件及差异基因筛选标准
| 类型 | 软件 | 标准化方法 | pvalue计算模型 | FDR计算方法 | 差异基因筛选标准 |
|---|---|---|---|---|---|
| 有生物学重复 | DESeq2(Anders et al, 2014) | DESeq | 负二项分布 | BH | |log2(FoldChange)| > 0 & padj < 0.05 |
| 无生物学重复 | edgeR(Robinson et al, 2010) | TMM | 负二项分布 | BH | |log2(FoldChange)| > 1 & padj < 0.05 |
若按照以上标准筛选得到的差异基因过少(低于100),很有可能导致后面的功能富集分析没有显著性结果,所以,我们会根据项目的具体情况,适当地降低筛选差异基因的阈值标准。若项目实验只关注某几个基因的表达情况(如基因敲除),不在意富集结果,从下面的差异分析表格中筛选关注的那几个基因即可。
一般来说,如果一个基因在两组样品中的表达量差异达到两倍以上,我们认为这样的基因是具有表达差异的。为了判断两个样品之间的表达量差异究竟是由于各种误差导致的还是本质差异,我们需要对所有基因在这两个样本中的表达量数据进行假设检验。而转录组分析是针对成千上万个基因进行的,这样会导致假阳性的累积,基因数目越多,假设检验的假阳性累积程度会越高,所以引入padj对假设检验的P-value进行校正,从而控制假阳性的比例[3]。
差异基因的筛选标准是非常重要的,我们给出的标准|log2(FoldChange)| > 1 & padj< 0.05是常用的经验值,在实际项目中可以根据情况灵活选择。例如,差异倍数可以选择1.5倍,也可以选择3倍,padj常用的阈值包括0.01、0.05、0.1等。若按照以上标准筛选得到的差异基因过少,很有可能导致后⾯的功能富集分析没有显著性结果。若项目实验只关注某几个基因的表达情况(如基因敲除),不在意富集结果,从下面的差异分析表格中筛选关注的那几个基因即可。反之,如果得到的差异基因数目过多,不利于后续目标基因的筛选,这个时候可使用更严格的阈值标准进行筛选,则可以使用更严格的阈值标准进行筛选。
2.6.1. 差异miRNA的筛选
通过Deseq2进行差异分析,我们通常采用 |log2FC|>1 & padj < 0.05 进行差异miRNA的筛选,差异计算结果如下。
结果文件:
| 结果说明 | 结果文件 |
|---|---|
| 定量分析表达矩阵结果 | 04.DE/miR.Counts.csv |
| 差异miRNA分析结果(ALL) | 04.DE/Allgene_anno_ALL.xls |
| 差异miRNA分析结果(筛选后) | 04.DE/Allgene_anno.xls |
04.DE/Allgene_anno.xls表头
| 表头 | 说明 |
|---|---|
ENSEMBL | 差异miRNA的ENSEMBL名 |
pvalue | 差异miRNA的置信度计算结果 |
padj | 差异miRNA的多重校验FDR |
log2FC | Treat组 vs Control组 差异倍数 的log2标准化结果 |
FC | Treat组 vs Control组 差异倍数 |
log2FC_abs | Treat组 vs Control组 差异倍数 的log2标准化结果的绝对值(此列便于筛选log2FC阈值) |
FC_HvsL | 高表达组 vs 低表达组 差异倍数 (此列便于筛选FC阈值) |
change | 使用本次分析的阈值,对差异miRNA的上下调标记 |
miRNA | 差异miRNA的miRNA名 |
ENTREZID | 差异miRNA的ENTREZID号 |
GENENAME | 差异miRNA的基本描述信息 |
baseMean | 差异miRNA的表达量标准化后的平均值 |
Samples* | 样本的原始表达矩阵表达量结果 |
Samples*_normal | 样本的表达矩阵标准化后的结果 |
2.6.2. 差异miRNA的热图聚类
将所有比较组的差异miRNA取并集之后作为差异miRNA集。两组以上的实验,可对差异miRNA集进行聚类分析,将表达模式相近的基因聚在一起。我们采用主流的层次聚类对基因的表达值进行聚类分析,对行(row)进行均一化处理(Z-score)。热图中表达模式相近的基因或样本会被聚集在一起,每个方格中的颜色反映的不是基因表达值,而是表达数据的行进行均一化处理后得到的数值(一般在-1到1之间),所以热图中的颜色只能横向比较(同一基因在不同样本中的表达情况),不能纵向比较(同一样本不同基因的表达情况)。结果文件中既有组间的聚类,也有样品间的聚类。结题报告展示了样品间的聚类,具体如下图所示。

图 2.6.2. 差异表达基因聚类热图
图中横坐标为样品名,纵坐标为差异miRNA归一化后的数值,颜色越红,表达量越高,越蓝,表达量越低。
结果文件:
差异miRNA的热图结果:
04.DE/heatmap/
2.6.3. 差异miRNA的火山图分布
火山图可直观展示每个比较组合的差异miRNA分布情况,如下图所示。图中横坐标表示基因在处理和对照两组中的表达倍数变化(log2FoldChange),纵坐标表示基因在处理和对照两组中表达差异的显著性水平(-log10padj或-log10pvalue)。为上调基因用红色点表示,下调基因用蓝色点表示。

图 2.6.3. 差异miRNA火山图
图中横坐标为log2FoldChange值,纵坐标为-log10padj或-log10pvalue,蓝色的虚线表示差异基因筛选标准的阈值线
结果文件:
差异基因的火山图结果:
04.DE/volcano/
2.7. 靶基因预测
miRNA是一类在生物体内起到重要调控作用的的小片段非编码RNA。一般认为miRNA通过和mRNA的结合,可以抑制mRNA的表达,从而影响到Gene的表达。
2.7.1. 靶基因预测结果
目前miRNA的靶基因预测软件主要是依据miRNA与其靶位点的互补性、miRNA与其靶位点的调控关系、miRNA靶位点的保守性、miRNA-mRNA双链之间的热稳定性及附近序列的二级结构等原则设计的。 目前miRNA预测主要基于算法预测,目前也有一部分以实验结果为收集对象的数据库。使用算法预测的数据库,有如 TargetScan、miRDB、RNAhybrid、DIANA-microT-CDS 等。以实验实验验证的收集的靶标相互作用数据库,有如 miRTarBase、Tarbase v8 等。 考虑到研究时效性,我们选择基于miRbase22为基础研究的数据库,在此基础上筛选出近5年内较新的数据库。
其中,miRDB数据库 是根据 “miRNA通过下调其靶基因的表达” miRNA主要功能特征,采用机器学习来进行模型预测得到的靶标数据库。它通过采用大量RNA数据集,使用 “过表达的miRNA“ 及 “下调的转录本” 进一步量化miRNA靶向下调的特征,采用SVM算法开发了预测模型 MirTarget。 该算法结合实验数据同时比较 TargetScan,DIANA-MicroT,miRanda 和 PITA 另外四种预测算法,获得了最优表现。 该模型对于每个候选靶标基因,它都会生成由SVM计算出的概率分数,该分数反映了预测结果的统计置信度,分数越高,置信度越高。所有预测目标的目标预测分数在50到100之间,一般而言,预测得分 > 80 的预测目标具有较高的可信度。如果分数低于60,则需要谨慎选择,一般需要提供其他证据支持。
TargetScan数据库 则基于序列互补原则,找到比对到靶 3'UTR 的保守性 8 mer、7 mer 或 6 mer 位点(seed match 序列),进一步根据热力学稳定性筛选得到 miRNA 的靶基因。 随后,对于不具备保守性的 seed match 区域计算相应的 context score。 将保守和不保守区域的 context score 进行排序即得到 context score percentile。 一般考虑 context score percentile > 90 为预测的可能具有功能的 miRNA 的靶基因。
综上,miRDB数据库主要通过miRNA-mRNA调控关系来进行量化计算,而TargetScan通过序列互补原则筛选后再进行热稳定性量化计算,前者相较于后者更有说服力,而后者得到的数据较为全面。我们综合两者,通过标准 miRDB Score >= 80 & TargetScan context score percentile >= 90 的靶基因作为预测结果。通过预测给出的miRNA-靶基因对,用于后续进一步网络图或富集分析。
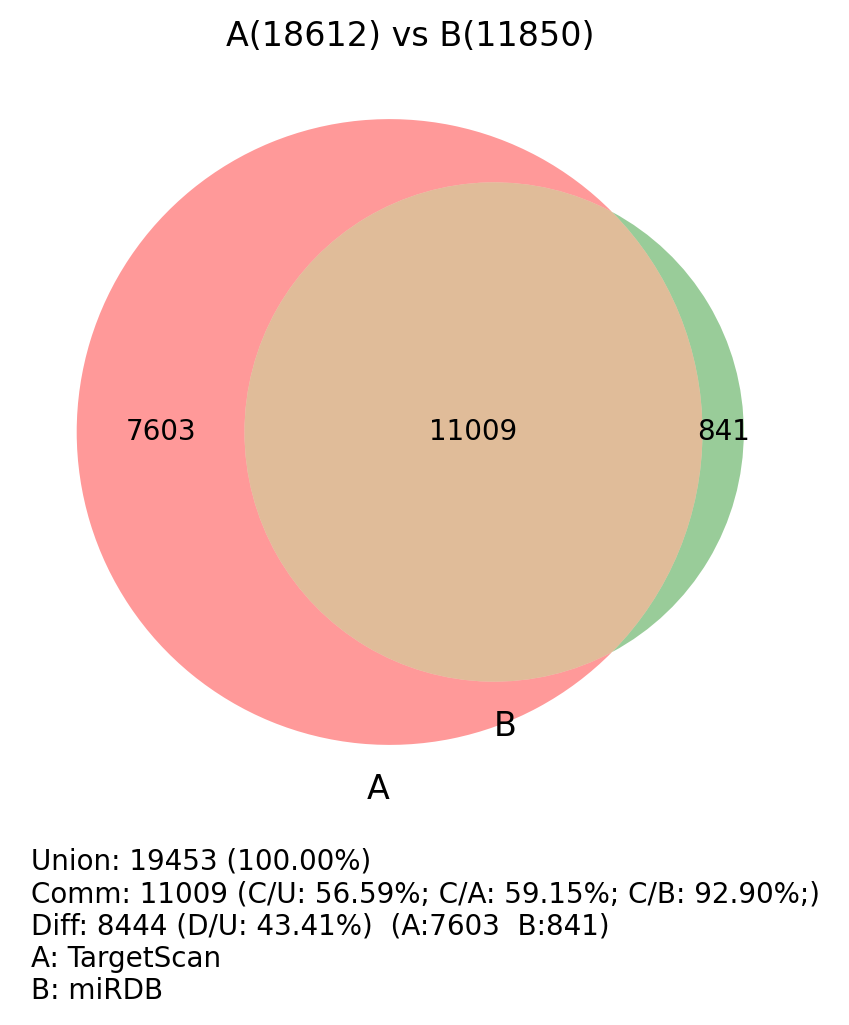
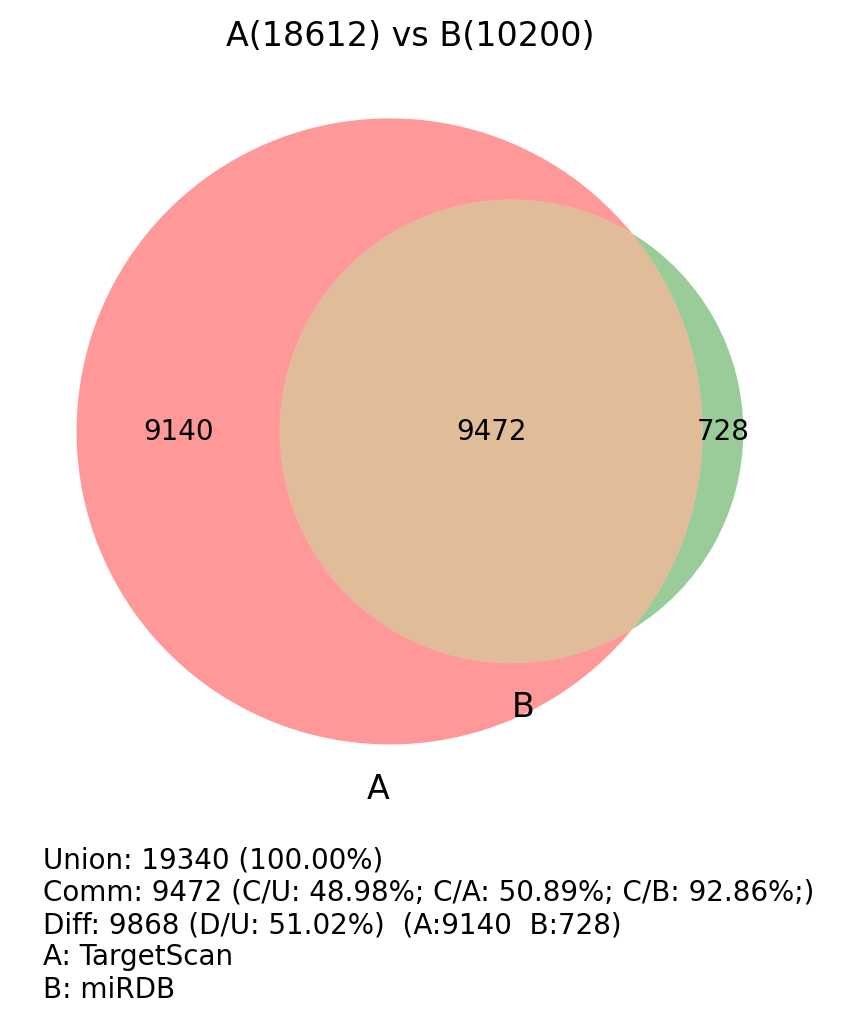
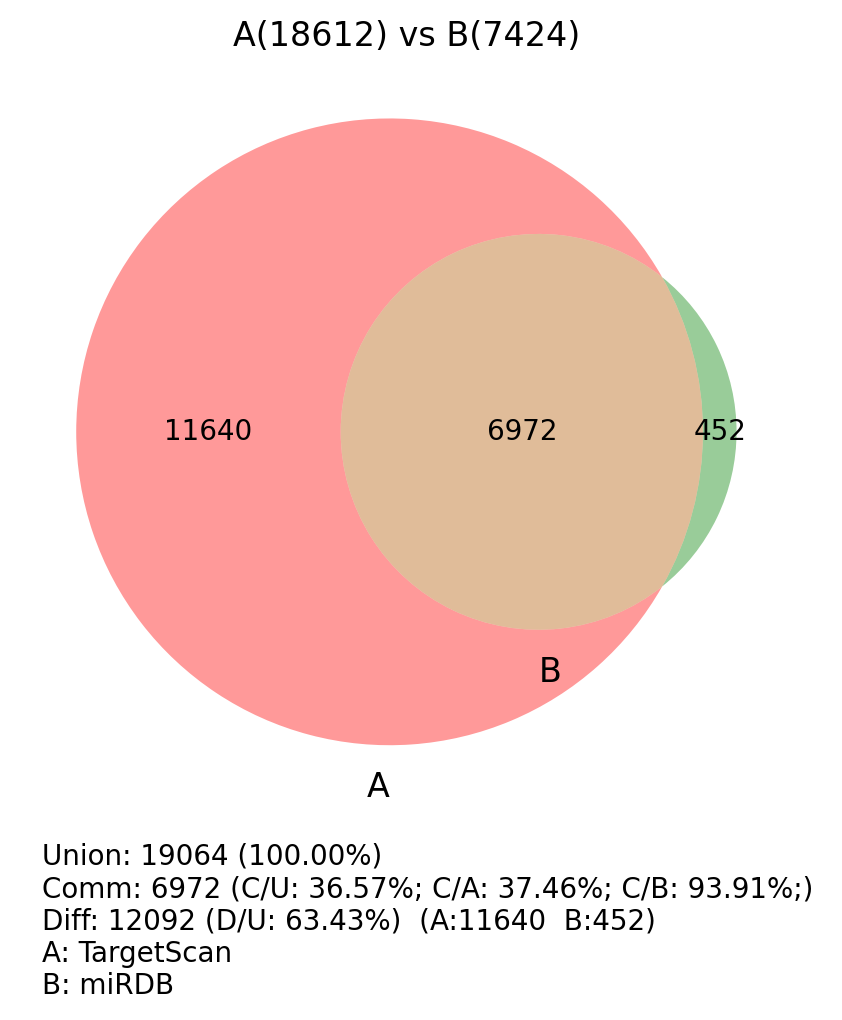
图 2.7.1. TargetScan vs miRDB 的靶基因交集veen图
结果文件:
miRDB/TargetScan预测结果:
结果说明 结果文件 miRDB预测结果(原始) 05.Target/*/Res_miRDB/Res_*.xlsmiRDB预测结果( miRDB Score >= 80过滤后)05.Target/*/Res_miRDB/Res_*.sig.xlsmiRDB预测结果(过滤情况统计) 05.Target/*/Res_miRDB/Res_*.Summary.xlsTargetScan预测结果(原始) 05.Target/*/Res_TargetScan/Res_*.xlsTargetScan预测结果( TargetScan score >= 90过滤后)05.Target/*/Res_TargetScan/Res_*.sig.xlsTargetScan预测结果(过滤情况统计) 05.Target/*/Res_TargetScan/Res_*.Summary.xlsmiRDB/TargetScan预测结果过滤后的交集结果:
结果说明 结果文件 miRDB/TargetScan两者过滤后预测结果交集统计(总表) 05.Target/*/Compare/Compare_diffMirna2_ALL.xlsmiRDB/TargetScan两者过滤后预测结果交集统计(仅共存的)
(当两者中出现仅有一个软件有结果时,则按一个软件筛选)05.Target/*/Compare/Compare_diffMirna2.xls仅共存列表的各个miRNA的靶基因数量统计 05.Target/*/Compare/Compare_Summary_miRNA.xls仅共存列表的各个靶基因的相应miRNA数量统计 05.Target/*/Compare/Compare_Summary_SYMBOL.xls交集veen统计文件夹 05.Target/*/Compare/veen
以上结果均位于文件夹: 05.Target/
Results/05.Target/Compare/veen/ 交集veen统计文件夹说明
| 表头 | 说明 |
|---|---|
| 所有基因 | |
| 比较组A和B的所有基因 | compare/union0_all_* |
| 比较组A的基因 | compare/union1_A_* |
| 比较组B的基因 | compare/union2_B_* |
| 共有基因 | |
| 比较组的共有基因 | compare/com_all_* |
| 特有基因 | |
| 比较组A和B的特有基因 | compare/diff0_all_* |
| 比较组A的特有基因 | compare/diff1_A_* |
| 比较组B的特有基因 | compare/diff2_B_* |
| 比较组A和B的veen图及相关说明 | compare/venn_gene_* |
2.8. 富集分析
我们根据基因表达量分析得到差异基因之后,必须进一步落到基因的功能上来。对于转录组分析而言,往往涉及到成千上万个基因,这会使分析变得很复杂。解决思路是将一个基因列表分成多个部分,从而减少分析的复杂度。为了解决怎么分成不同类,通常会对基因功能进行富集分析, 期望发现在生物学过程中起关键作用的生物通路, 从而揭示和理解生物学过程的基本分子机制。功能富集分析可以将成百上千个基因、蛋白或者其他分子分到不同的通路中,以减少分析的复杂度。另外,在两种不同实验条件下,激活的通路显然比简单的基因或蛋白列表更有说服力。基因功能富集分析首先要构建基因集(gene set,如GO和KEGG数据库等),也就是基因组注释信息进行分类。然后再把我们的目标基因集(差异基因集或者其他基因集)映射到背景基因集上,注意区分注释与富集。
我们采用clusterProfiler软件对差异基因集进行GO功能富集分析,KEGG通路富集分析等。富集分析基于超几何分布原理,其中差异基因集为差异显著分析所得差异基因并注释到GO或KEGG数据库的基因集,背景基因集为所有进行差异显著分析的基因并注释到GO或KEGG数据库的基因集。富集分析结果是对每个差异比较组合的所有差异基因集、上调差异基因集、下调差异基因集进行富集。本报告中展示的表格是选取某一个比较组合的富集分析结果,图片是所有组合的富集分析结果。

图 2.8.1. 基因富集分析原理图
2.8.1. 富集分析结果文件
| 结果说明 | 结果路径 |
|---|---|
| GO富集分析结果 | |
| GO富集结果列表(所有结果) | 06.ENRICH/*/gene.ego_all-p.adjust1.00.csv |
| GO富集结果列表(按p.adj<0.05筛选后) | 06.ENRICH/*/gene.ego_all-p.adjust0.05.csv |
| GO富集结果列表(MF、BP、CC所有结果) | 06.ENRICH/*/gene.ego_ALL.csv |
| GO富集分析柱状图 | 06.ENRICH/*/gene.GO-*-barplot.p* |
| GO富集分析散点图 | 06.ENRICH/*/gene.GO-*-dotplot.p* |
| GO富集分析DAG图 | 06.ENRICH/*/gene.GO-*-DAG.p* |
| KEGG富集分析结果 | |
| KEGG富集结果列表(所有) | 06.ENRICH/*/gene.KEGG.csv |
| KEGG富集结果列表(按p.adj<0.05筛选后) | 06.ENRICH/*/gene.KEGG_significant.csv |
| KEGG富集分析柱状图 | 06.ENRICH/*/gene.KEGG-*-barplot.p* |
| KEGG富集分析散点图 | 06.ENRICH/*/gene.KEGG-*-dotplot.p* |
结果文件夹:
分析结果文件夹:
06.ENRICH/以上富集分析各图的可视化网页:
all-pdf.html、up-pdf.html、down-pdf.html
表头说明: (Results/*enrich_*/gene.ego_*.csv GO富集结果列表)
| ID | 对应GO数据库中的ID |
|---|---|
| ONTOLOGY | 分子功能(Molecular Function),生物过程(biological process)和细胞组成(cellular component) |
| Description | GO的描述 |
| GeneRatio | 对应GO 差异基因数 / 能够对应到GO数据库中同类型的差异基因数 |
| BgRatio | 对应GO包含对应物种的基因数 / GO数据库中包含对应物种的基因数 |
| pvalue | 富集分析得到的p-value |
| p.adjust | 校正后的p-value |
| qvalue | 富集分析得到的qvalue |
| Count | 富集基因数目 |
| ENTREZID | 富集基因列表(ENTREZID) |
| SYMBOL | 富集基因列表(SYMBOL) |
表头说明: (Results/*enrich_*/gene.KEGG*.csv KEGG富集结果列表)
| ID | 对应PATHWAY数据库中的ID |
|---|---|
| Description | PATHWAY的描述 |
| GeneRatio | 对应PATHWAY 差异基因数 / 能够对应到PATHWAY数据库中的差异基因数 |
| BgRatio | 对应PATHWAY包含对应物种的基因数 / PATHWAY数据库中包含对应物种的基因数 |
| pvalue | 富集分析得到的p-value |
| p.adjust | 校正后的p-value |
| qvalue | 富集分析得到的qvalue |
| Count | 富集基因数目 |
| ENTREZID | 富集基因列表(ENTREZID) |
| SYMBOL | 富集基因列表(SYMBOL) |
2.8.2. GO功能富集分析
GO(Gene Ontology)是描述基因功能的综合性数据库,可分为生物过程(biological process)和细胞组成(cellular component)分子功能(Molecular Function)三个部分。GO功能富集以padj小于0.05作为为显著性富集的阈值,富集结果见结果文件。
从GO富集分析结果中,选取最显著的30个Term绘制柱状图进行展示,若不足30个,则绘制所有Term,按生物过程、细胞组分和分子功能三大类别及差异基因上下调分类画的柱状图。
有向无环图(Directed Acyclic Graph,DAG)为差异基因GO富集分析结果的图形化展示方式。图中,分支代表包含关系,从上至下所定义的功能范围越来越小,选取每个差异比较组合的GO富集结果最显著性前5位的GO Term作为有向无环图的主节点,并通过包含关系,将相关联的GO Term一起展示,颜色的深浅代表富集程度。我们的项目中分别绘制生物过程、分子功能和细胞组分的DAG图。

图 2.8.2.1. GO富集分析柱状图
图中纵坐标为GO Term,横坐标为GO Term富集的显著性水平,数值越高越显著

图 2.8.2.2. GO富集分析散点图
图中横坐标为注释到GO Term上的差异基因数与差异基因总数的比值,纵坐标为GO Term

图 2.8.2.3. GO富集分析DAG图
每个节点代表一个GO术语,方框代表的是富集程度为TOP5的GO,颜色的深浅代表富集程度,颜色越深就表示富集程度越高,每个节点上展示了该TERM的名称及富集分析的padj
2.8.3. KEGG通路富集分析
KEGG(Kyoto Encyclopedia of Genes and Genomes)是整合了基因组、化学和系统功能信息的综合性数据库。KEGG通路富集以padj小于0.05作为显著性富集的阈值,富集结果见结果文件。
从KEGG富集结果中,选取最显著的20个KEGG通路绘制柱状图进行展示,若不足20个,则绘制所有通路,如下图所示。图中横坐标为通路富集的显著性水平,数值越高越显著,纵坐标为KEGG通路。
从KEGG富集结果中,选取最显著的20个KEGG通路绘制散点图进行展示,若不足20个,则绘制所有通路,如下图所示。图中横坐标为注释到KEGG通路上的差异基因数与差异基因总数的比值,纵坐标为KEGG通路,点的大小代表注释到KEGG通路上的基因数,颜色从红到紫代表富集的显著性大小。

图 2.8.3.1. KEGG富集分析柱状图
图中横坐标为通路富集的显著性水平,数值越高越显著,纵坐标为KEGG通路。

图 2.8.3.2. KEGG富集散点图
图中横坐标为注释到KEGG通路上的差异基因数与差异基因总数的比值,纵坐标为KEGG通路