![]()
广州市黄埔区学大道揽月路广州企业孵化器B座402
电话:020-85625352
手机:18102256923、18102253682
Email:servers@gzscbio.com
Fax:020-85625352
QQ:386244141
项目名称:motif结果能给到我们些什么信息?
所属分类:生物信息学分析
联系电话:020-85625352
QQ:386244141
Email:servers@gzscbio.com
技术服务描述
motif结果能给到我们些什么信息?
1. 背景简介
1.1. 什么是motif?
Motif是一段典型的序列或者一个结构。一般来说,我们称为基序。一般情况下是指构成任何一种特征序列的基本结构。通俗来讲,即是有特征的短序列,一般认为它是拥有生物学功能的保守序列,可能包含特异性的结合位点,或者是涉及某一个特定生物学过程的有共性的序列区段。比如蛋白质的序列特异性结合位点,如核酸酶和转录因子。
1.2. 研究motif的意义
序列基序在基因调控分析中越来越重要。Motif并不局限于基因组DNA序列还是RNA序列,甚至蛋白质序列也可以提取出相应的motif。通过序列信息的解析,我们可以解析生物学过程中的密码。当某一个大类的序列中,大量重复出现相同的序列结构的时候,就可能存在探索它意义的价值。基于motif序列的提取,我们可以预测潜在的结合位点等等,有助于我们进一步理解各生物学过程中涉及的生物学意义。比如转录因子的结合位点,其motif往往意味着某蛋白结构域与DNA碱基序列的相互作用。
1.3. motif的展示形式
在我们的日常应用中,我们经常会看到motif这个词的出现,往往会伴随着这样一张logo展示图。这样的logo经常用于描述序列特征,如DNA中的蛋白质结合位点等。motif logo由每个位置的一堆字母组成。字母的相对大小表示它们在序列中的频率。每个字母的高度与该位置的相应碱基的出现频率成正比,常以bits为单位。每个位置的字母按照保守性从大到小排列,可以方便的从顶端的字母识别保守序列。

图1 logo展示图
用一段序列来描述所有序列的碱基组成,称之为一致性序列。当一个位置涉及到多个碱基出现的时候,这里就涉及到简并碱基的使用。
示例如图2,在日常使用中,也经常用“RRACH”来表示它。这个“RRACH”就是一种一致性序列的表示方式。
| 简并碱基 | 正常碱基 |
|---|---|
| R | A/G |
| Y | C/T |
| M | A/C |
| K | G/T |
| S | G/C |
| W | A/T |
| H | A/T/C |
| B | G/T/C |
| V | G/A/C |
| D | G/A/T |
| N | A/T/C/G |

图2 logo示例
2. motif如何被预测?
HOMER 是一套用于Motif查找和二代数据分析的工具。它不仅能检测已知的motif,还能预测可能存在的未知motif。 这个预测新颖motif算法,设计用于在基因组学应用里的调控元件分析(这里的元件指DNA,而非蛋白质)。 它采取两套序列,并试图识别在其中一套序列上相对于另一套的特定富集的调控元件(比如说我们的peaks序列相对于整个基因组序列)。它使用ZOOPS评分(0或1发生在每条序列上)与超几何富集计算(或二项式)来确定motif富集。HOMER也尽力考虑数据集里的排序偏差。它的设计用于ChIP-Seq和启动子分析,但可以应用于几乎任何核酸序列的motif发现。
我们使用 Homer 子程序 findMotifsGenome.pl 进行motif分析, findMotifsGenome.pl 命令用于在基因组区域中寻找富集Motifs。以下为结果的详细解读。
3. 结果列表说明
Demo: motif_ZYN-H3K27ac - Homer Known Motif Enrichment Results
结果说明:
| 结果 | 说明 |
|---|---|
homerMotifs.all.motifs | 由所有homerMotifs.motifs*组成的连接文件 |
homerMotifs.motifs10 | de novo 查找motif的输出文件(motif长度为10) |
homerMotifs.motifs12 | de novo 查找motif的输出文件(motif长度为12) |
homerMotifs.motifs8 | de novo 查找motif的输出文件(motif长度为8) |
homerResults.html | 查找已知motifs的格式化输出。 |
homerResults/ | 子文件夹:包含homerResults.html网页文件, 包括motif<#>.motif文件,用于寻找每个motif的具体实例。 |
knownResults.html | de novo查找motif的格式化输出。 |
knownResults/ | 子文件夹:包含knownResults.html网页文件, 包括known<#>.motif文件。 |
knownResults.txt | 包含关于已知motifs丰富的统计信息的文本文件(在EXCEL中打开)。 |
motifFindingParameters.txt | 用于执行findMotifsGenome.pl的命令,包含分析使用的参数 |
seq.autonorm.tsv | 用于lower-order oligo标准化的autonormalization统计。 |
3.1. 网页可视化文件的表头说明
3.1.1. knownResults.html (Homer Known Motif Enrichment Results)
图示:
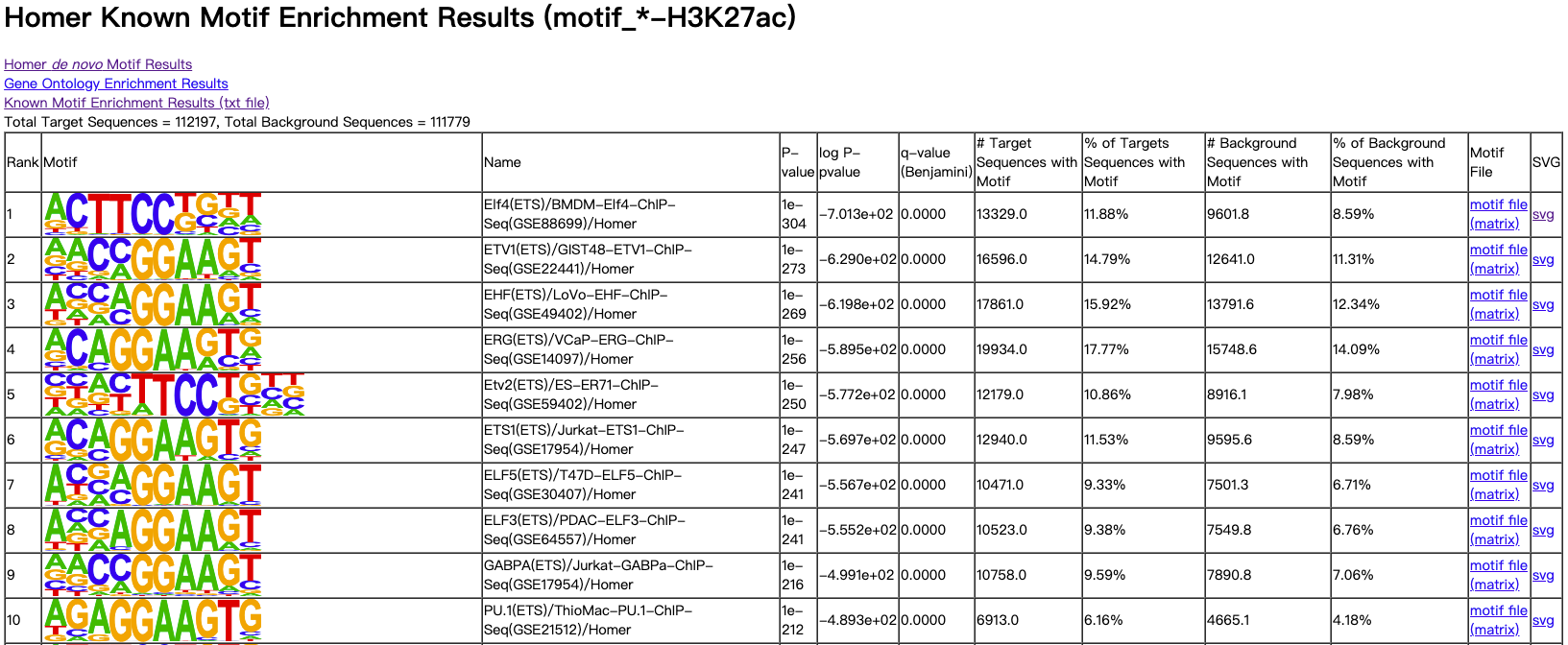
表头说明:
| 表头 | 说明 |
|---|---|
Rank | 序号 |
Motif | motif序列可视化 |
P-value | 置信度计算结果 |
log P-pvalue | 计算log10P值结果 |
q-value (Benjamini) | Benjamini模型计算的q值 |
# Target Sequences with Motif | 靶标序列 |
% of Targets Sequences with Motif | 靶标序列占总序列百分比 |
# Background Sequences with Motif | 背景序列 |
% of Background Sequences with Motif | 背景序列占总序列百分比 |
Motif File | motif文件结果 |
SVG | motif的svg可视化文件 |
3.1.2. homerResults.html (Homer de novo Motif Results)
图示:
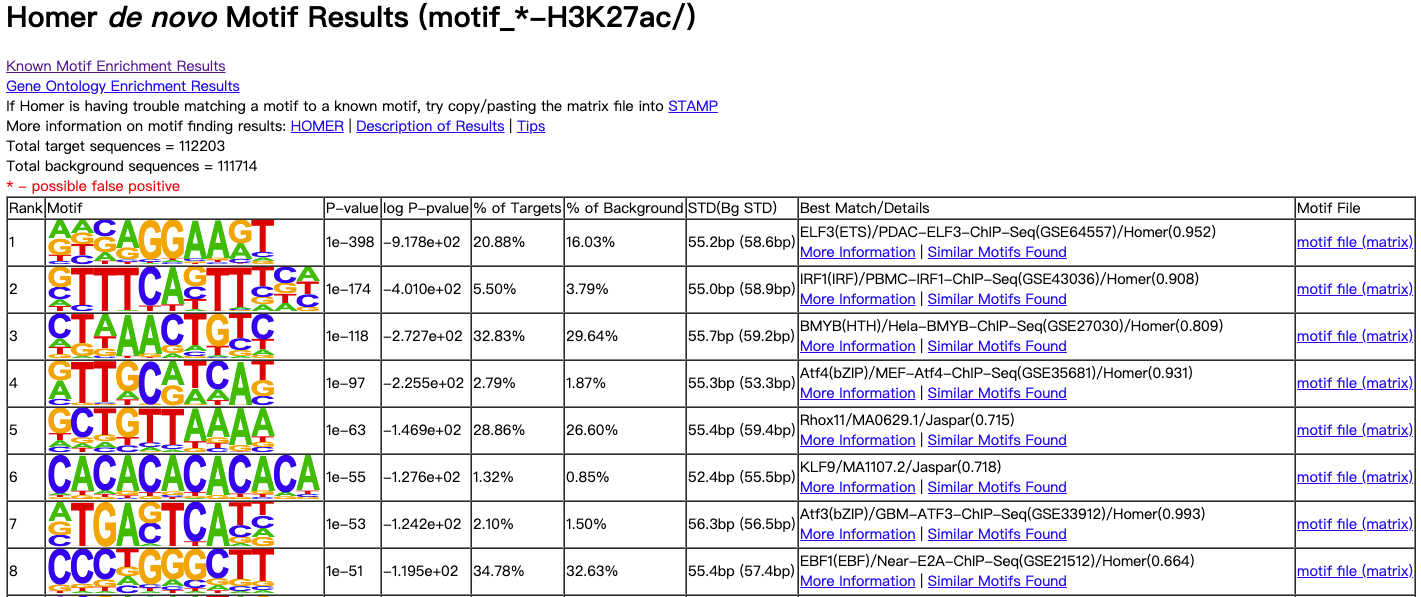
表头说明:
| 表头 | 说明 |
|---|---|
Rank | 序号 |
Motif | motif序列可视化 |
P-value | 置信度计算结果 |
log P-pvalue | log P-pvalue 值 |
% of Targets | 靶标序列占总denovo序列百分比 |
% of Background | 背景序列占总denovo序列百分比 |
STD(Bg STD) | 靶标和背景的序列集出现偏离序列中心200bp的标准偏差 |
Best Match/Details | 最为匹配的结果 |
Motif File | motif文件结果 |
3.2. 如理解输出的Motif文件homerMotifs.*.motifs* ?
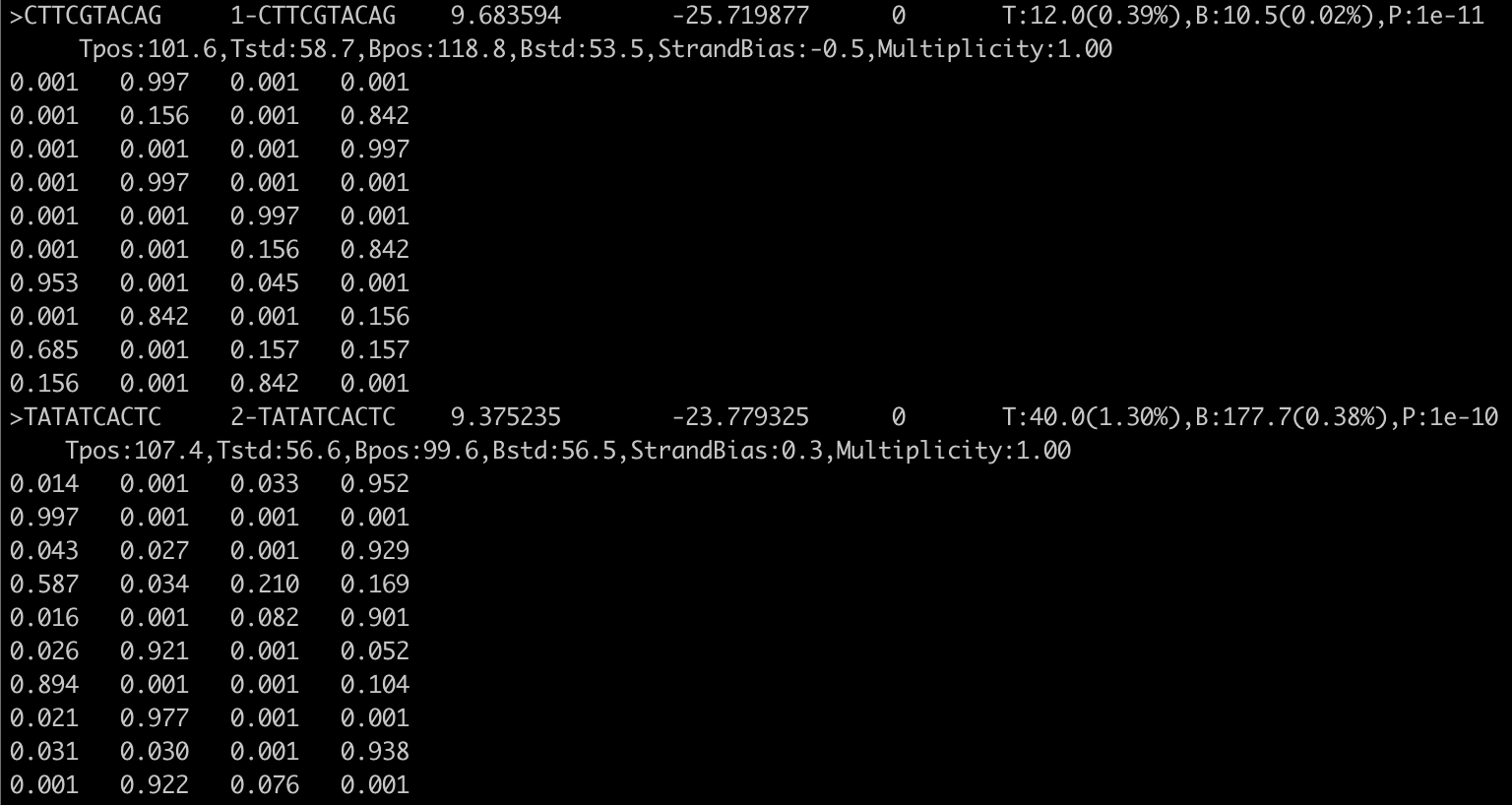
第一行以一个“>”开头,后面跟着各种信息,其他行是每个位置的各个核苷酸具体概率(A/C/G/T)。标题行实际上是用制表符分隔的,并包含以下信息:
1. “>”+序列(实际上不用于任何东西,可以是空的)示例:>NNATGASTCATH
2. motif名称(如果几个motif在同一个文件中,应该是唯一的)。例子:Fra1(bZIP)/BT549-Fra1-ChIP-Seq(GSE46166)/Homer
3. log odds检测阈值,用于确定结合的vs未结合位点。示例:7.011739
4. 富集的log-p值
5. 0(表示向后兼容性的占位符,在旧版本中用于描述“gapped” motif,结果证明它并不是很有用)
6. 发生的信息,用逗号分隔,例如: T:4.0(57.14%),B:3353.3(3.23%),P:1e-4
- T: 带motif的靶标序列数,占靶序列总数的%
- B: 带motif的背景序列数目,占总背景的%
- P: 最终富集P值
7. 用逗号分隔的Motif统计信息(这一个信息是与链有关的,因为我的BED文件里链的哪一列没有指定是+/-,我输入的是.,所以这里没有第7个信息),官网的例子:Tpos:100.7,Tstd:32.6,Bpos:100.1,Bstd:64.6,StrandBias:0.0,Multiplicity:1.13
- Tpos: motif在目标序列中的平均位置(0 =序列开始)
- Tstd: 目标序列中位置的标准偏差
- Bpos: motif在背景序列中的平均位置(0 =序列开始)
- Bstd: 背景序列中位置的标准差
- StrandBias: 在+链出现与在-链出现的log比例。
- Multiplicity: 在具有一个或多个结合位点的序列中,每个序列平均出现的次数。